
ONUREG 300 mg, comprimé pelliculé, flacon de 14
Retiré du marché le : 20/09/2021
Dernière révision : 07/06/2021
Taux de TVA : 0%
Laboratoire exploitant : CELGENE
Source :

ONUREG est indiqué pour le traitement de maintenance chez les patients adultes atteints de leucémie aiguë myéloïde (LAM) ayant obtenu une rémission complète (RC) ou une rémission complète avec récupération incomplète de la numération formule sanguine (RCi) après une thérapie d'induction avec ou sans traitement de consolidation, et qui ne sont pas éligibles à une greffe de cellules souches hématopoïétiques (GCSH).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients. Allaitement (voir rubrique Fertilité, grossesse et allaitement).
Toxicité hématologique
Le traitement par ONUREG peut être associé à une neutropénie, une thrombocytopénie et une neutropénie fébrile (voir rubrique Effets indésirables pour les fréquences). L'interruption, la réduction ou l'arrêt d'ONUREG peuvent être nécessaires pour gérer les toxicités hématologiques. Les patients doivent être avertis de signaler rapidement les épisodes fébriles. Les patients dont la numération plaquettaire est basse doivent être avertis de signaler les premiers signes ou symptômes de saignement. Des soins de support tels que des antibiotiques et / ou des antipyrétiques pour la prise en charge de l'infection/fièvre et du GCSF pour la neutropénie doivent être prescrits en fonction des caractéristiques individuelles du patient, de la réponse au traitement et conformément aux recommandations cliniques actuelles (voir rubrique Posologie et mode d'administration Tableau 1).
Toxicité gastro-intestinale
Les toxicités gastro-intestinales ont été les effets indésirables les plus fréquents chez les patients traités par ONUREG (voir rubrique Effets indésirables). Les patients doivent recevoir un traitement antiémétique prophylactique pendant les 2 premiers cycles de traitement par ONUREG (voir rubrique Posologie et mode d'administration). La diarrhée doit être traitée rapidement dès l'apparition des symptômes. L'interruption, la réduction ou l'arrêt d'ONUREG peuvent être nécessaires pour gérer les toxicités gastro-intestinales (voir rubrique Posologie et mode d'administration).
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et jusqu'à 6 mois après l'arrêt du traitement. Les hommes doivent utiliser une contraception efficace pendant le traitement et jusqu'à 3 mois après l'arrêt du traitement (voir rubrique Fertilité, grossesse et allaitement).
Intolérance au lactose
Les comprimés d'ONUREG contiennent du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c'est à dire qu'il est essentiellement «sans sodium».
Résumé du profil de sécurité
Les effets indésirables les plus fréquents sont nausées (64,8%), vomissements (59,7%), diarrhée (50,4%), neutropénie (44,5%), fatigue / asthénie (44,1%)5, constipation (38,6%), thrombocytopénie (33,5%), douleurs abdominales (21,6%)4, infection des voies respiratoires (17%)2, arthralgie (13,6%), diminution de l'appétit (12,7%), neutropénie fébrile (11,9%), douleur dorsale (11,9%), leucopénie (10,6%), douleur aux extrémités (10,6%) et pneumonie (10,2%)1.
Des effets indésirables (EIs) graves sont survenus chez 16,1% des patients recevant ONUREG. Les effets indésirables graves les plus courants sont la neutropénie fébrile (6,8%) et la pneumonie (5,1%)1.
Un arrêt définitif d'ONUREG en raison d'un effet indésirable est survenu chez 6,8% des patients. Les EIs les plus courants nécessitant un arrêt définitif sont les nausées (2,1%), la diarrhée (1,7%) et les vomissements (1,3%).
Des interruptions de dose dues à un effet indésirable sont survenues chez 36,4% des patients ayant reçu ONUREG. Les EIs nécessitant une interruption de dose comprennent la neutropénie (19,9%), la thrombocytopénie (8,5%), les nausées (5,5%), la diarrhée (4,2%), les vomissements (3,8%), la pneumonie (3,4%)1, la leucopénie (2,5%), la neutropénie fébrile (2,1%) et douleurs abdominales (2,1%)4.
Des réductions de dose dues à une période d'effets indésirables sont survenues chez 14% des patients ayant reçu ONUREG. Les effets indésirables nécessitant une réduction de la posologie comprenaient la neutropénie (5,5%), la diarrhée (3,4%), la thrombocytopénie (1,7%) et les nausées (1,7%).Liste tabulée des effets indésirables
Le tableau 2 présente les effets indésirables par classe de système d'organes (SOC) et par fréquence rapportés dans l'étude pivot de phase 3 avec ONUREG. Au total, 236 patients ont reçu ONUREG. La durée médiane du traitement était de 11,6 mois (intervalle: 0,5 à 74,3 mois) pour le bras ONUREG.
Les fréquences sont définies selon la convention MedDRA: très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à <1/10); peu fréquent (≥ 1/1 000 à <1/100); rare (≥ 1/10 000 à <1/1 000); très rare (<1/10 000); fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. Les effets indésirables sont présentés dans le tableau ci-dessous selon la fréquence la plus élevée observée.
Tableau 2 : Effets indésirables (EI) chez les patients atteints de LAM recevant un traitement de maintenance par ONUREG
| Classe de système d'organes | Fréquence, tous grades confondusa |
| Infections et infestations | Très fréquent Pneumonie1, 6, infection des voies respiratoires 2 Fréquent Grippe, infection urinaires3, bronchite, rhinite |
| Affections hématologiques et du système lymphatique | Très fréquent Neutropénie, thrombocytopénie6, neutropénie fébrile6, leucopénie |
| Troubles du métabolisme et de la nutrition | Très fréquent Diminution de l'appétit |
| Affections psychiatriques | Fréquent Anxiété |
| Affections gastro-intestinales | Très fréquent Nausées, vomissements, diarrhée, constipation, douleurs abdominales4 |
| Affections musculo-squelettiques et du tissu conjonctif | Très fréquent Arthralgie, douleur dorsale, douleur aux extrémités |
| Troubles généraux et anomalies au site d'administration | Très fréquent Fatigue / asthénie5 |
| Investigations | Fréquent Perte de poids |
a Tous les EIs avec au moins 5,0% des patients dans le bras ONUREG et au moins 2,0% plus fréquents que dans le bras placebo.
1 Termes groupés incluant pneumonie, aspergillose bronchopulmonaire, infection pulmonaire, pneumonie à Pneumocystis jirovecii, pneumonie atypique, pneumonie bactérienne et pneumonie fongique.
2 Termes groupés incluant infections des voies respiratoires supérieures*, infections des voies respiratoires* et infections des voies respiratoires virales*.
3 Termes groupés incluant les infections des voies urinaires*, infections des voies urinaires bactériennes*, infections des voies urinaires à Escherichia et cystite.
4 Termes groupés incluant douleurs abdominales, douleurs abdominales hautes, inconforts abdominaux et douleurs gastro- intestinales.
5 Termes groupés incluant fatigue et asthénie.
6 Effets indésirables dont au moins un a été considéré comme menaçant le pronostic vital (si le résultat de la réaction était le décès, il est inclus dans les cas de décès).
Description de certains effets indésirables
Toxicité hématologique
Une neutropénie nouvelle ou aggravée de grades 3 ou supérieurs (41,1%), une thrombocytopénie (22,5%) ou une neutropénie fébrile (11,4%) ont été fréquemment rapportées chez les patients traités par ONUREG. La première survenue de neutropénie, thrombocytopénie ou neutropénie fébrile de grades 3 ou 4 est survenue au cours des 2 premiers cycles chez 19,9%, 10,6% et 1,7% respectivement chez les patients traités par ONUREG. Voir la rubrique Posologie et mode d'administration pour les recommandations de surveillance et de prise en charge.
Toxicité gastro-intestinale
Les toxicités gastro-intestinales ont été les effets indésirables les plus fréquents chez les patients traités par ONUREG. Des nausées (64,8%), des vomissements (59,7%) et des diarrhées (50,4%) ont été rapportés chez des patients traités par ONUREG. Une diarrhée de grade 3 ou plus est survenue chez 5,1% des patients et des vomissements et des nausées de grade 3 ou plus sont survenus respectivement chez 3,0% et 2,5% des patients traités par ONUREG. La première occurrence de nausées, de vomissements ou de diarrhée de grade 3 ou 4 est survenue au cours des 2 premiers cycles chez 1,7%, 3,0% et 1,3%, respectivement, chez les patients traités par ONUREG. Voir rubrique
4.2 pour les recommandations de surveillance et de prise en charge.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté à l'aide de la fiche de déclaration des effets indésirables disponible dans le Protocole d'utilisation thérapeutique et de recueil d'informations.
PROPHYLAXIE : Les patients doivent être traités avec un antiémétique 30 minutes avant chaque dose d'azacitidinepour les 2 premiers cycles de traitement. La prophylaxie antiémétique peut être arrêtée après 2 cycles, s'il n'y a pas eu de nausées, ni de vomissements.
SURVEILLANCE du traitement
:
NFS : avant traitement, toutes les deux semaines pendant les 2 premiers cycles (56 jours), toutes les deux semaines pendant les 2 cycles suivants l'adaptation posologique, et tous les mois par la suite, avant le début des cycles de traitement suivants.
INFORMER les patients masculins et féminins sur la possibilité de cryoconservation du sperme ou de l'ovule avant de commencer le traitement.
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et jusqu'à 6 mois après l'arrêt du traitement. Les hommes doivent être avisés de ne pas concevoir d'enfant pendant le traitement et doivent utiliser une contraception efficace pendant le traitement et jusqu'à 3 mois après l'arrêt du traitement (voir rubriques Mises en garde et précautions d'emploi et Données de sécurité précliniques).
Grossesse
Il n'existe pas de données suffisantes sur l'utilisation d'ONUREG chez la femme enceinte. Des études chez la souris et le rat ont montré une toxicité pour la reproduction et le développement (voir rubrique Données de sécurité précliniques). Le risque potentiel chez l'humain est inconnu. Sur la base des résultats d'études animales et de son mécanisme d'action, ONUREG n'est pas recommandé pendant la grossesse (en particulier pendant le premier trimestre, sauf en cas de nécessité absolue) et chez les femmes en âge de procréer n'utilisant pas de contraception. Les bénéfices du traitement doivent être mis en balance avec le risque éventuel pour le foetus dans chaque cas individuel. Si une patiente ou une partenaire tombe enceinte pendant qu'elle prend ONUREG, la patiente doit être informée du risque potentiel pour le foetus.
Allaitement
L'excrétion de l'azacitidine ou ses métabolites dans le lait maternel n'est pas connue. En raison des effets indésirables potentiels graves chez l'enfant allaité, l'allaitement est contre-indiqué pendant le traitement par ONUREG (voir rubrique Contre-indications).
Fertilité
Il n'y a pas de données chez l'humain sur l'effet de l'azacitidine sur la fertilité. Chez les animaux, les effets indésirables de l'azacitidine sur la fertilité des mâles ont été documentés (voir rubrique Données de sécurité précliniques). Les patients qui souhaitent concevoir un enfant doivent être invités à consulter un expert en matière de reproduction et de cryoconservation de l'ovule ou du sperme avant de commencer le traitement par ONUREG.
Aucune étude clinique formelle d'interaction médicamenteuse avec l'azacitidine n'a été menée.
En cas d'administration concomitante avec d'autres agents antinéoplasiques, la prudence et la surveillance sont recommandées car un effet pharmacodynamique antagoniste, additif ou synergique ne peut être exclu. Ces effets peuvent dépendre de la dose, de la fréquence et du schéma d'administration.
L'exposition à ONUREG était peu affectée lors de la co-administration avec un inhibiteur de la pompe à protons (oméprazole). Par conséquent, une modification de la dose n'est pas nécessaire lorsque ONUREG est co-administré avec des inhibiteurs de la pompe à protons ou d'autres modificateurs de pH.
Une étude in vitro de l'azacitidine avec des fractions hépatiques humaines a indiqué que l'azacitidine n'était pas métabolisée par les isoformes du cytochrome P450 (CYP). Par conséquent, les interactions avec les inducteurs ou les inhibiteurs du CYP sont considérées comme peu probables (voir rubrique Propriétés pharmacocinétiques).
Les effets inhibiteurs ou inductifs cliniquement significatifs de l'azacitidine sur le métabolisme des substrats du cytochrome P450 sont peu probables (voir rubrique Propriétés pharmacocinétiques). Aucune interaction médicamenteuse cliniquement pertinente n'est attendue lorsqu' ONUREG est co-administré avec des substrats de la glycoprotéine P (P-gp), de la protéine de résistance au cancer du sein (BCRP), des transporteurs organiques d'anions (OAT) OAT1 et OAT3, des polypeptides de transport d'anions organiques (OATP) OATP1B1 et OATP1B3, ou transporteur de cations organiques (OCT) OCT2.
L'azacitidine n'est pas un substrat de la P-gp, on ne s'attend donc pas à ce qu'elle interagisse avec les inducteurs ou inhibiteurs de la P-gp.
ONUREG doit être initié et suivi sous la supervision d'un médecin expérimenté dans l'utilisation de de médicaments de chimiothérapie.
Les patients doivent être traités avec un antiémétique 30 minutes avant chaque dose d'ONUREG pour les 2 premiers cycles de traitement. La prophylaxie antiémétique peut être arrêtée après 2 cycles, s'il n'y a pas eu de nausées, ni de vomissements (voir rubrique Mises en garde et précautions d'emploi).
Posologie
La dose recommandée est de 300 mg d'azacitidine par voie orale une fois par jour. Chaque cycle consiste en une période de traitement de 14 jours suivie d'une période sans traitement de 14 jours (cycle de traitement de 28 jours).
ONUREG doit être poursuivi jusqu'à ce qu'un maximum de 15 % de blastes soit observé dans le sang périphérique ou la moelle osseuse, ou jusqu'à ce que la toxicité ne soit plus acceptable (voir les modalités de modification du schéma posologique en cas de rechute de la maladie).
ONUREG ne doit pas être utilisé de manière substituable avec l'azacitidine injectable en raison des différences d'exposition, de dose et de calendrier de traitement. Il est recommandé aux professionnels de la santé de vérifier le nom du médicament, la dose et la voie d'administration.
Tests de laboratoire
Une numération formule sanguine complète doit être effectuée avant le début du traitement. Une surveillance complète de la numération formule sanguine est également recommandée toutes les deux semaines pendant les 2 premiers cycles (56 jours), toutes les deux semaines pendant les2 cycles suivants l'adaptation posologique, et tous les mois par la suite, avant le début des cycles de traitement suivants (voir rubrique Mises en garde et précautions d'emploi).
Modification du schéma posologique en cas de rechute de la LAM
En cas de rechute de la maladie, avec 5% à 15% de blastes dans le sang périphérique ou la moelle osseuse, en conjonction avec une évaluation clinique, une extension du schéma posologique de 14 à 21 jours de cycles répétés de 28 jours doit être envisagée. Le traitement ne doit pas dépasser 21 jours pendant toute période de 28 jours.
ONUREG doit être arrêté si plus de 15% de blastes sont observés dans le sang périphérique ou la moelle osseuse ou à la discrétion du médecin.
Adaptation de la dose pour effets indésirables
Les modalités d'adaptation de dose pour effets indésirables hématologiques et non hématologiques sont recommandées en fonction des évaluations cliniques et des résultats du laboratoire (voir tableau 1).
Tableau 1: Adaptations posologiques pour effets indésirables hématologiques et non hématologiques
| Critères* | Action recommandée |
| Neutropénie de grade 4 ou Neutropénie de grade 3 avec fièvre | Première occurrence · Interrompre ONUREG. Reprenez le cycle de traitement à la même dose une fois que la neutropénie revient aux grades 2 ou inférieurs. · Utiliser des soins de support tels que le facteur de croissance des colonies de granulocytes (GCSF), comme indiqué cliniquement (voir rubrique 4.4). Occurrence lors de 2 cycles consécutifs · Interrompre ONUREG. Reprenez le cycle de traitement à une dose réduite de 200 mg après le retour de la neutropénie au grade 2 ou inférieur. · Si un patient continue à présenter une toxicité après une réduction de dose, réduisez la durée du traitement de 7 jours. · Si la toxicité persiste ou réapparaît après la réduction de la dose et de la durée de traitement, arrêtez ONUREG. · Utiliser des soins de support tels que le GCSF, selon le tableau clinique (voir rubrique 4.4). |
| Thrombocytopénie de grade 4 ou Thrombocytopénie de grade 3 avec saignements | Première occurrence · Interrompre ONUREG. Reprenez le cycle de traitement à la même dose une fois que la thrombocytopénie revient au grade 2 ou inférieur. Occurrence lors de 2 cycles consécutifs · Interrompre ONUREG. Reprenez le cycle de traitement à une dose réduite de 200 mg après le retour de la thrombocytopénie au grade 2 ou inférieur. · Si un patient continue à présenter la toxicité après une réduction de dose, réduisez la durée du traitement de 7 jours. · Si la toxicité persiste ou réapparaît après la réduction de la dose et de la durée de traitement, arrêtez ONUREG. |
| Nausées, vomissements ou diarrhée de Grade 3 ou plus | · Interrompre ONUREG. Reprenez le cycle de traitement à la même dose une fois que la toxicité est revenue au grade 1 ou ou inférieur. · Utilisez des soins de support tels qu'un traitement antiémétique et traitez la diarrhée dès l'apparition des symptômes (voir rubrique 4.4). · Si l'événement se reproduit, interrompre la dose jusqu'à ce qu'il soit résolu au grade 1 ou inférieur et réduire la dose à 200 mg. · Si un patient continue à présenter la toxicité après une réduction de dose, réduisez la durée du traitement de 7 jours. |
| | · Si la toxicité persiste ou réapparaît après la réduction de la dose et de la durée de traitement, arrêtez ONUREG. |
| Autres événements non hématologiques de grade 3 ou plus | · Interrompre ONUREG et fournir des soins médicaux selon les recommandations locales. Reprenez le cycle de traitement à la même dose une fois que la toxicité est revenue au grade 1 ou inférieur. · Si la toxicité réapparaît, interrompez ONUREG jusqu'à ce que celle-ci soit revenue au grade 1 ou inférieur et réduisez la dose à 200 mg. · Si un patient continue à présenter la toxicité après une réduction de dose, réduisez la durée du traitement de 7 jours. · Si la toxicité persiste ou réapparaît après la réduction de la dose et de la durée de traitement, arrêtez ONUREG. |
*Le grade 1 est léger, le grade 2 est modéré, le grade 3 est sévère, le grade 4 engage le pronostic vital.
Ces grades de toxicité sont conformes à la terminologie commune du National Cancer Institute pour les événements indésirables version 4.3 (NCI-CTCAE v4.3).
Doses manquées ou retardées
Si une dose d'ONUREG est oubliée ou si elle n'est pas prise à l'heure habituelle, la dose doit être prise dès que possible le même jour. Ensuite, la prochaine dose programmée doit être prise à l'heure habituelle le jour suivant. Deux doses ne doivent pas être prises le même jour.
Si une dose est vomie, une autre dose ne doit pas être prise le même jour. La dose doit être prise à l'heure habituelle d'administration, le jour suivant.
Populations particulières
Patients âgés
Aucun ajustement posologique n'est recommandé chez les patients de plus de 65 ans (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
ONUREG peut être administré aux patients présentant une insuffisance rénale légère, modérée ou sévère sans ajustement posologique initial (voir rubrique Propriétés pharmacocinétiques). Les patients seront surveillés pour les effets indésirables et la dose sera modifiée au besoin.
Insuffisance hépatique
Aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance hépatique légère (bilirubine totale (BIL) ≤ limite supérieure de la normale (LSN) et aspartate aminotransférase (AST)> LSN, ou BIL 1 à 1,5 × LSN et n'importe quelle valeur d'AST) (voir rubrique Propriétés pharmacocinétiques).
Les patients présentant une insuffisance hépatique modérée (BIL> 1,5 à 3 × LSN) et sévère (BIL> 3 × LSN) doivent être surveillés plus fréquemment pour déceler des effets indésirables et un ajustement posologique approprié doit être effectué (voir Tableau 1).
Population pédiatrique
La sécurité et l'efficacité d'ONUREG chez les enfants et adolescents de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
ONUREG est à usage oral.
ONUREG peut être pris avec ou sans nourriture.
Les comprimés doivent être avalés entiers avec un verre d'eau à peu près à la même heure chaque jour. Ils ne doivent pas être coupés, écrasés, dissous ou mâchés (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination).
Durée de conservation :
30 mois.
Précautions particulières de conservation :Conserver les flacons entre 20 ° C et 25 ° C; transports autorisés entre 15 ° C et 30 ° C. Garder le flacon bien fermé. Conserver et distribuer dans le flacon d'origine (avec deux capsules de dessiccant).
Sans objet.
En cas de surdosage, le patient doit être surveillé avec une numération de la formule sanguine appropriée et un traitement d'appoint doit être fourni, si nécessaire, conformément aux recommandations locales. Il n'y a pas d'antidote spécifique connu en cas de surdosage avec ONUREG.
Classe pharmacothérapeutique : Agents antinéoplasiques, antimétabolites, analogues de la pyrimidine, code ATC: L01BC07.
Mécanisme d'action
L'azacitidine est un inhibiteur de l'ADN méthyltransférase et un modificateur épigénétique. L'azacitidine est incorporée dans l'ADN et l'ARN après absorption cellulaire et biotransformation enzymatique en nucléotides triphosphates. L'incorporation de l'azacitidine dans l'ADN des cellules leucémiques induit une modification des voies épigénétiques par inhibition des ADN méthyltransférases, et réduit la méthylation de l'ADN. Cela a conduit à une modification de l'expression des gènes, y compris la réexpression des gènes régulant la suppression des tumeurs, les voies immunitaires, le cycle cellulaire et la différenciation cellulaire. L'incorporation d'azacitidine dans l'ARN des cellules LAM, inhibe l'ARN méthyltransférase, réduit la méthylation de l'ARN, diminue la stabilité de l'ARN et diminue la synthèse des protéines.
Efficacité et sécurité cliniques
L'efficacité et la tolérance d'ONUREG ont été étudiées dans une étude de phase 3 multicentrique randomisée contrôlée par placebo, Quazar AML-001 (CC-486-AML-001) en double aveugle et en groupes parallèles, évaluant ONUREG versus placebo comme traitement de maintenance chez les patients atteints de LAM. Les patients ont été recrutés avec une LAM de novo, une LAM secondaire à un diagnostic antérieur de syndrome myélodysplasique (SMD) ou une leucémie myélomonocytaire chronique (LMMC); les patients étaient âgés de ≥ 55 ans et avaient obtenu une première rémission complète (RC) ou une rémission complète avec récupération incomplète de la formule sanguine (RCi) dans les 4 mois (+ / - 7 jours) après une chimiothérapie d'induction intensive avec ou sans traitement de consolidation. Les patients n'étaient pas éligibles pour la GCSH au moment de la randomisation, qui comprenait des patients qui n'avaient pas de donneur de greffon, ou qui avaient choisi de ne pas procéder à une GCSH.
Les patients des deux bras de l'étude ont reçu les meilleurs soins de support jugés nécessaires par l'investigateur. Les meilleurs soins de support comprenaient, mais sans s'y limiter, un traitement par transfusions de globules rouges (GR), des transfusions de plaquettes, une utilisation d'un agent stimulant l'érythropoïèse, un traitement antibiotique, antiviral et / ou antifongique, GCSF, un traitement antiémétique et un support nutritionnel.Les patients ayant obtenu un RC/RCi après la fin du traitement d'induction intensif avec ou sans consolidation ont reçu ONUREG 300 mg (N = 236) ou un placebo (N = 233) une fois par jour les jours 1 à 14 de chaque cycle de 28 jours. En cas de rechute de la maladie (5% à 15% de blastes dans le sang périphérique ou la moelle osseuse), le schéma posologique a été étendu à 21 jours de cycles de traitement répétés de 28 jours à la discrétion du médecin. Le traitement s'est poursuivi jusqu'à progression de la maladie (plus de 15% de blastes ont été observés dans le sang périphérique ou la moelle osseuse) ou jusqu'à une toxicité inacceptable.
Un total de 472 patients ont été randomisés 1:1 entre les bras de traitement ONUREG et placebo. Les caractéristiques démographiques et pathologiques initiales de la population de patients atteints de LAM étaient équilibrées entre les bras de traitement, comme indiqué dans le tableau 3. La durée médiane du traitement était de 11,6 mois (intervalle: 0,5 à 74,3 mois) pour le bras ONUREG versus 5,7 mois (intervalle: 0,7 à 68,5 mois) pour le bras placebo. Un total de 51 patients (21%) recevant ONUREG et 40 patients (17%) recevant le placebo ont étendu leur schéma posologique à 300 mg par jour pendant 21 jours en raison d'une rechute de la LAM.
Sur les 469 patients de l'étude de phase 3 qui ont reçu un traitement, 61% (285/469) étaient âgés de 65 ans ou plus et 11% (51/469) avaient 75 ans ou plus. Aucune différence globale de tolérance ou d'efficacité d'ONUREG n'a été observée entre ces patients et les patients plus jeunes.
Tableau 3: Données démographiques de base et caractéristiques liées à la maladie dans l'étude CC-486-AML-001
| Paramètre | ONUREG (N = 238) | Placebo (N = 234) |
| Age (ans) | | |
| Médiane (min, max) | 68,0 (55, 86) | 68,0 (55, 82) |
| Catégories d'âge n (%) | | |
| <65 ans | 66 (27,7) | 68 (29,1) |
| ≥65 ans to <75 ans | 144 (60,5) | 142 (60,7) |
| ≥75 ans | 28 (11,8) | 24 (10,3) |
| Sexe, n (%) | | |
| Hommes | 118 (49,6) | 127 (54,3) |
| Femmes | 120 (50,4) | 107 (45,7) |
| Origine ethno géographique, n (%) | | |
| Caucasien | 216 (90,8) | 197 (84,2) |
| Noir ou Afro-Americain | 2 (0,8) | 6 (2,6) |
| Asiatique | 6 (2,5) | 20 (8,5) |
| Autre | 12 (5,0) | 11 (4,7) |
| Non collecté or reporté | 2 (0,8) | 0 (0) |
| Score de Performance ECOG, n (%) | | |
| 0 | 116 (48,7) | 111 (47,4) |
| 1 | 101 (42,4) | 106 (45,3) |
| 2 | 21 (8,8) | 15 (6,4) |
| 3 | 0 (0) | 2 (0,9) |
| Risque Cytogénétique au diagnostic, n (%) | | |
| Risque intermediare1 | 203 (85,3) | 203 (86,6) |
| Risque défavorable2 | 35 (14,7) | 31 (13,2) |
| Classification Initiale de la LAM, n (%) | | |
| LAM avec anomalies génétiques récurrentes | 39 (16,4) | 46 (19,7) |
| LAM avec des changements liés à la myélodysplasie | 49 (20,6) | 42 (17,9) |
| Paramètre | ONUREG (N = 238) | Placebo (N = 234) |
| Néoplasmes myéloïdes liés au traitement | 2 (0,8) | 0 (0) |
| Autre LAM | 148 (62.2) | 145 (62,0) |
| Donnée manquante | 0 (0) | 1 (0,4) |
| Type de LAM, n (%) | | |
| Primaire (de novo) | 213 (89,5) | 216 (92,3) |
| Secondaire | 25 (10,5) | 18 (7,7) |
| Maladie résiduelle (MRD) au moment de la randomisation3, n (%) | | |
| Négative | 133 (55,9) | 111 (47,4) |
| Positive | 103 (43,3) | 116 (49,6) |
| Donnée manquante | 2 (0,8) | 7 (3,0) |
LAM=Leucémie Aigue Myéloïde, ECOG=Eastern Cooperative Oncology Group, MRD=Minimal Residual Disease
1 Unrisque intermédiairea été défini comme une cytogénétique normale +8, t (9; 11) ou autre non défini.
2 Un risque défavorable a été défini comme complexe (≥ 3 anomalies): -5; 5q-; -7; 7q-; 11q23 - non t (9; 11); inv (3); t (3; 3); t (6; 9); ou t (9; 22). Source pour les risques intermédiaires et défavorable : National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology for AML.
3Le statut de la maladie résiduelle (MRD) dans la moelle osseuse a été mesuré pendant la période de dépistage par un test cytométrique en flux à un niveau de sensibilité de 0,1%.
La plupart des patients ont reçu un traitement de consolidation après le traitement d'induction dans les bras de traitement ONUREG (78%) et placebo (82%) ; plus de 90% de ces patients dans chaque bras de traitement ont reçu 1 ou 2 cycles de traitement de consolidation après le traitement d'induction (tableau 4).
Tableau 4: Traitement de consolidation dans l'étude CC-486-AML-001
| Paramètre | ONUREG (N=238) | Placebo (N=234) |
| A reçu un traitement de consolidation après l'induction | | |
| OUI, n (%) | 186 (78,2) | 192 (82,1) |
| 1 Cycle, n (%) | 110 (46,2) | 102 (43,6) |
| 2 Cycles, n (%) | 70 (29,4) | 77 (32,9) |
| 3 Cycles, n (%) | 6 (2,5) | 13 (5,6) |
| No, n (%) | 52 (21,8) | 42 (17,9) |
| RC / RCi au moment de la randomisation | | |
| RC, n (%) | 183 (76,9) | 177 (75,6) |
| RCi, n (%) | 50 (21,0) | 44 (18,8) |
| Non RC/RCi a, n (%) | 5 (2,1) | 11 (4,7) |
| Donnée manquante, n (%) | 0 (0) | 2 (0,9) |
RC = rémission complète; RCi = RC morphologique avec récupération incomplète de la formule sanguine.
a Ces patients avaient une moelle osseuse initiale avec moins de 5% de blastes et à la fois un nombre absolu de neutrophiles <1 x 109 et des plaquettes <100 x 109.
L'efficacité d'ONUREG chez les patients adultes atteints de LAM a été établie sur la base de la survie globale (SG) et de la survie sans rechute (SSR).
Les résultats d'efficacité sont résumés dans le tableau 5.
Tableau 5: Résultats d'efficacité de CC-486-AML-001 (population ITT)
| Critères d'efficacité | ONUREG (N=238) | Placebo (N=234) |
| Survie globale | | |
| Evènement de SG, n (%) | 158 (66,4) | 171 (73,1) |
| SG médiane (95% IC) mois | 24,7 (18,7 ; 30,5) | 14.8 (11,7 ; 17,6) |
| Critères d'efficacité | ONUREG (N=238) | Placebo (N=234) |
| Hazard ratio (95% IC) p value | 0,69 (0,55 ; 0,86) 0,0009 | |
| Survie sans rechute | | |
| Evènement, n (%) | 164 (68,9) | 181 (77,4) |
| SSR médiane (95% IC) mois | 10,2 (7,9 ; 12,9) | 4,8 (4,6 ; 6,4) |
| Hazard ratio (95% IC) p value | 0,65 (0,52 ; 0,81) 0,0001 | |
| Délai de rechute | | |
| Ayant rechuté, n (%) | 154 (64,7) | 179 (76,5) |
| Délai de rechute médian mois (95% IC) | 10,2 (8,3 ; 13,4) | 4,9 (4,6 ; 6,4) |
| Délai avant l'arrêt du traitement | | |
| Traitement interrompu, n (%) | 193 (81,1) | 208 (88,9) |
| Délai avant l'arrêt du traitement mois (95% IC) | 11,4 (9,8 ; 13,6) | 6,1 (5,1 ; 7,4) |
| Traitement interrompu - rechute de la maladie, n (%) | 143 (60,1) | 180 (76,9) |
IC = Intervalle de confiance.
Des analyses de sous-groupes prédéfinis de la SG et de la SSR ont montré un effet de traitement homogène pour ONUREG dans les sous-groupes démographiques et liés à la maladie, y compris le risque cytogénétique de base, le nombre de cycles de consolidation antérieurs reçus et le statut RC / RCi.
Les courbes de Kaplan-Meier affichent les résultats de la SG (voir Figure 1) et de la SSR (voir Figure 2).
Figure 1: Courbe de Kaplan-Meier pour la survie globale: ONUREG versus placebo (population ITT)
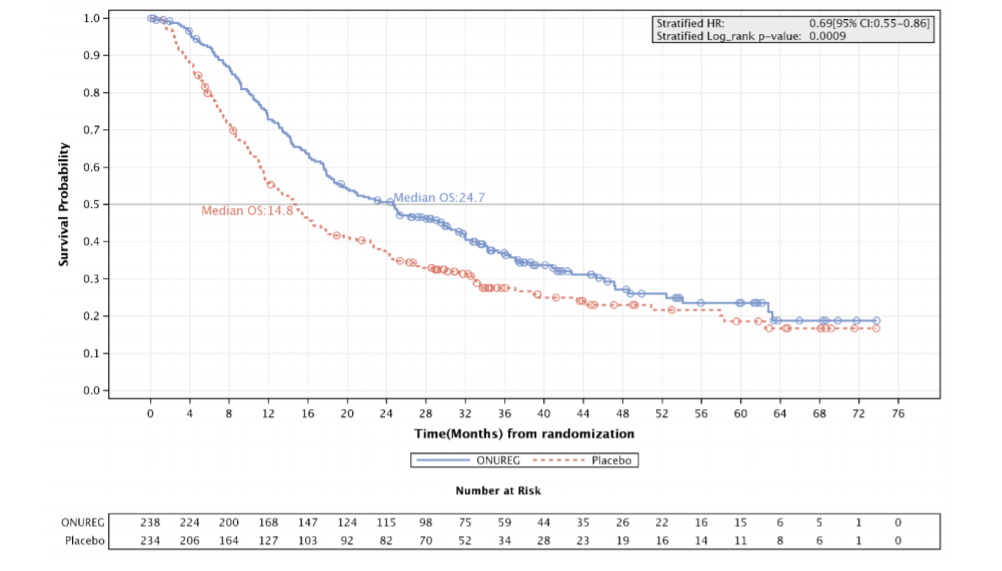
Figure 2: Courbe de Kaplan-Meier pour la survie sans rechute: ONUREG versus placebo (population ITT)

Chez les patients dont le schéma posologique a été prolongé à 300 mg pendant 21 jours en raison d'une rechute de la maladie, la SG médiane (22,8 mois pour ONUREG et 14,6 mois pour le placebo) et la SSR médiane (7,4 mois pour ONUREG et 4,6 mois pour le placebo) étaient comparables aux résultats globaux de l'étude.
ONUREG a démontré un effet thérapeutique favorable sur la SG par rapport au placebo chez les patients présentant une maladie résiduelle minimale (MRD) et les patients avec une MRD négative. L'effet du traitement sur la SG était plus prononcé chez les patients MRD-positifs (HR = 0,69; IC à 95%: 0,51, 0,93) comparé à celui des patients MRD négatifs (HR = 0,81; IC à 95%: 0,59, 1,12).
Qualité de vie liée à la santé (Health Related Quality of Life- HRQoL)
La HRQoL a été évaluée à l'aide de l'échelle d'évaluation fonctionnelle FACIT (Functional Assessment of Chronic Illness Therapy-fatigue scale - échelle de fatigue), de l'indice d'utilité de la santé à cinq dimensions à trois niveaux (EQ-5D-3L) et d'une échelle visuelle analogique (EVA). Au départ, les patients présentaient un faible niveau de fatigue et un bon niveau de HRQoL qui étaient généralement comparables à ceux de la population générale d'âge similaire. Ce niveau de HRQoL a été maintenu au fil du temps avec ONUREG, par rapport à la valeur initiale, ainsi qu'avec le placebo. Le délai de détérioration définitive et la proportion de patients présentant une détérioration cliniquement significative se sont avérés similaires entre ceux recevant ONUREG et le placebo. Dans l'ensemble, les résultats démontrent que la HRQoL était similaire entre le traitement par ONUREG et les bras placebo, sans détérioration cliniquement significative au fil du temps.
Absorption
L'exposition était généralement linéaire avec des augmentations proportionnelles à la dose de l'exposition systémique ; une forte variabilité inter-individuelle a été observée. Les valeurs moyennes géométriques (coefficient de variation [% CV]) Cmax et ASC après administration orale d'une dose unique de 300 mg étaient respectivement de 145,1 ng/mL (63,7) et 241,6 ng.h/mL (64,5). L'administration répétée du schéma posologique recommandé n'a pas entraîné d'accumulation de médicament. L'absorption de l'azacitidine a été rapide, avec un Tmax médian de 1 heure après l'administration. La biodisponibilité orale moyenne par rapport à l'administration sous-cutanée (SC) était d'environ 11%.
Effet de la nourritureL'impact de la nourriture sur l'exposition à ONUREG était minime. Par conséquent, ONUREG peut être administré avec ou sans nourriture.
Distribution
Après administration orale, le volume de distribution apparent moyen géométrique était de 12,6 L/kg pour une personne de 70 kg. La liaison de l'azacitidine aux protéines plasmatiques était de 6 à 12%.
Biotransformation
D'après les données in vitro, le métabolisme de l'azacitidine ne semble pas être médié par les isoenzymes du cytochrome P450 (CYP). L'azacitidine subit une hydrolyse spontanée et une désamination médiées par la cytidine désaminase.
Élimination
La clairance apparente moyenne géométrique était de 1 242 L/h et la demi-vie moyenne géométrique était d'environ 0,5 heure. Après l'administration intraveineuse d'azacitidine marquée au 14C à 5 patients cancéreux, l'excrétion urinaire cumulée était de 85% de la dose radioactive. L'excrétion fécale représentait <1% de la radioactivité administrée sur 3 jours. L'excrétion moyenne de radioactivité dans l'urine après l'administration sous-cutanée de 14C-azacitidine était de 50%. La quantité d'azacitidine inchangée récupérée dans l'urine par rapport à la dose était <2% après administration sous-cutanée (SC) ou orale. L'excrétion fécale n'a pas été mesurée après administration orale.
Effets pharmacodynamiques
L'effet régulateur épigénétique de l'azacitidine sur la réduction de la méthylation globale de l'ADN dans le sang a été maintenu avec une exposition prolongée de 300 mg par jour administrée pendant 14 ou 21 jours sur un cycle de 28 jours dans les cancers myéloïdes, y compris les patients atteints de LAM d'une étude de phase 1/2. Une corrélation positive a été observée entre l'exposition plasmatique à l'azacitidine et l'effet pharmacodynamique de la réduction de la méthylation globale de l'ADN dans le sang.
Populations particulières
Personnes âgées
Dans une analyse pharmacocinétique de population (PK) de 286 patients atteints de LAM, l'âge (46 à 93 ans) n'a pas eu d'effets cliniquement significatifs sur la pharmacocinétique d'ONUREG. Par conséquent, aucune modification posologique d ONUREG n'est requise, quel que soit l'âge du patient.
Insuffisance hépatique
Aucune étude formelle n'a été menée chez des patients présentant une insuffisance hépatique. Il est peu probable que l'insuffisance hépatique affecte la pharmacocinétique à un degré cliniquement pertinent car l'azacitidine subit une hydrolyse spontanée et une désamination médiées par la cytidine désaminase. Une analyse pharmacocinétique de population a déterminé que l'AST (8 à 155 U/L), l'ALT (5 à 185 U / L) et l'insuffisance hépatique légère (BIL ≤ LSN et AST> LSN, ou BIL 1 à 1,5 × LSN et n'importe quelle valeur d'AST) n'ont pas d'effets cliniquement significatifs sur la pharmacocinétique de l'azacitidine. Les effets d'une insuffisance hépatique modérée à sévère (BIL> 1,5 × LSN et n'importe quelle valeur d'AST) sur la pharmacocinétique de l'azacitidine sont inconnus.
Insuffisance rénale
Chez les patients atteints de cancer, la pharmacocinétique de l'azacitidine a été comparée chez 6 patients ayant une fonction rénale normale (CLcr> 80 mL / min) et 6 patients atteints d'insuffisance rénale sévère (CLcr <30 mL / min) après administration sous-cutanée quotidienne (jours 1 à 5 ) à 75 mg / m2 / jour. Une insuffisance rénale sévère a augmenté l'exposition à l'azacitidine d'environ 70% après une administration unique et de 41% après plusieurs administrations sous-cutanées. Cette augmentation de l'exposition n'était pas corrélée à une augmentation des événements indésirables. L'ajustement posologique n'est pas recommandé à l'initiation du traitement et les patients seront surveillés pour les effets indésirables et la dose sera modifiée au besoin.
Une analyse pharmacocinétique de population après une dose de 300 mg d'ONUREG a déterminé que les patients atteints d'insuffisance rénale légère (CLcr: ≥ 60 à <90 mL / min), modérée (CLcr: ≥30 à <60 mL / min) et sévère (CLcr: <30 mL / min), présentaient une augmentation de l'ASC plasmatique de l'azacitidine de 19%, 25% et 38%, respectivement. L'effet d'une insuffisance rénale sévère sur ONUREG était similaire à l'étude clinique sur l'insuffisance rénale mentionnée ci-dessus avec l'azacitidine injectable (augmentation d'environ 40% de l'ASC).
Origine ethno-géographique
Les effets de l'origine ethno-géographique sur la pharmacocinétique d'ONUREG sont inconnus.
ONUREG a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Une fatigue a été rapportée lors de l'utilisation d'ONUREG. Par conséquent, la prudence est recommandée lors de la conduite de véhicules ou de l'utilisation de machines.
Dans une étude de toxicité orale de 14 jours chez le chien, la mortalité est survenue aux doses de 8 et 16 mg / m2 / jour. La dose maximale tolérée (DMT) était de 4 mg / m2 / jour. À 1 dose ou à toutes les doses, une pancytopénie corrélée à une hypoplasie de la moelle osseuse, une déplétion lymphoïde, une dilatation des glandes / des lumières et une nécrose unicellulaire dans les cryptes muqueuses de l'intestin grêle et du gros intestin, et / ou une vacuolisation hépatocellulaire centro-lobulaire ont été observées. A la DMT, ces résultats ont été partiellement ou complètement résolus après 3 semaines. Après des administrations parentérales d'azacitidine à des doses comparables, une mortalité et des toxicités similaires pour certains organes cibles ont été observées chez les rongeurs, les chiens et les singes. Les données non cliniques issues d'études de toxicité à doses répétées avec l'azacitidine n'ont révélé aucun danger particulier pour l'homme.
L'azacitidine induit à la fois des mutations géniques et des aberrations chromosomiques dans les systèmes cellulaires bactériens et mammifères in vitro. La cancérogénicité potentielle de l'azacitidine a été évaluée chez la souris et le rat. L'azacitidine a induit des tumeurs du système hématopoïétique chez les souris femelles, lorsqu'elle est administrée par voie intrapéritonéale 3 fois par semaine pendant 52 semaines. Une incidence accrue de tumeurs dans le système lymphoréticulaire, les poumons, les glandes mammaires et la peau a été observée chez les souris traitées par l'azacitidine administrée par voie intrapéritonéale pendant 50 semaines. Une étude de tumorigénicité chez le rat a révélé une incidence accrue de tumeurs testiculaires.
Les premières études d'embryotoxicité chez la souris ont révélé une fréquence de 44% de décès embryonnaire intra-utérine (résorption accrue) après une seule injection intrapéritonéale d'azacitidine au cours de l'organogenèse. Des anomalies du développement dans le cerveau ont été détectées chez des souris ayant reçu de l'azacitidine au moment où avant la fermeture du palais dur. Chez le rat, l'azacitidine n'a provoqué aucun effet indésirable lorsqu'elle était administrée avant l'implantation, mais elle était clairement embryotoxique lorsqu'elle était administrée pendant l'organogenèse. Les anomalies foetales au cours de l'organogenèse chez le rat comprenaient: des anomalies du système nerveux central (SNC) (exencéphalie / encéphalocèle), des anomalies des membres (micromélie, pied bot, syndactylie, oligodactylie) et d'autres (microphtalmie, micrognathie, gastroschisis, oedème et anomalies des côtes).
L'administration d'azacitidine à des souris mâles avant l'accouplement avec des souris femelles non traitées a entraîné une diminution de la fertilité et une perte de progéniture au cours du développement embryonnaire et postnatal ultérieur. Le traitement des rats mâles a entraîné une diminution du poids des testicules et des épididymes, une diminution du nombre de spermatozoïdes, une diminution des taux de gestation, une augmentation des embryons anormaux et une augmentation de la perte d'embryons chez les femelles accouplées (voir rubrique Fertilité, grossesse et allaitement).
ONUREG est un médicament cytotoxique. Si la poudre du comprimés pelliculés d'ONUREG entre en contact avec la peau, la peau doit être lavée immédiatement et soigneusement avec de l'eau et du savon. Si la poudre entre en contact avec les muqueuses, la zone doit être soigneusement rincée à l'eau.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament soumis à prescription hospitalière.
Prescription hospitalière réservées aux hématologues et médecins compétents en maladie du sang.
Comprimé pelliculé.
ONUREG 300 mg comprimé pelliculé
Comprimé pelliculé brun, ovale, 19,0x9,0 mm, gravé «300» sur une face et «ONU» sur l'autre face.
Flacon en polyéthylène haute densité (PEHD) contenant deux capsules de dessiccant de gel de silice en PEHD et d'un bouchon de sécurité enfant.
Chaque flacon contient 14 comprimés pelliculés.
Azacitidine........................................................................................................................... 300 mg
ONUREG 300 mg comprimé pelliculé
Chaque comprimé pelliculé contient 300mg d'azacitidine.
Excipient(s) à effet notoire : Chaque comprimé d'ONUREG 300 mg contient 5,42 mg de lactose (sous forme de lactose monohydraté).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu du comprimé
Mannitol (E421)
Cellulose microcristalline silicifiée (E460, E551)
Croscarmellose sodique (E468)
Stéarate de magnésium (E572)
ONUREG 300 mg, pelliculage du comprimé
Opadry II marron contenant:
Hypromellose (E464)
Dioxyde de titane (E171)
Lactose monohydraté
Polyéthylène glycol/macrogols (E1521)Triacétine (E1518)
Oxyde de fer jaune (E172)
Oxyde de fer rouge (E172)
Oxyde de fer noir (E172).